SCT - Analysis and Modelling of Small Angle Scattering Data
SCT is a suite of software tools designed to aid the comparison of molecular models of biological systems to small angle scattering data.
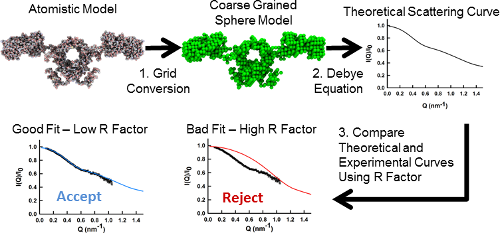
Small angle X-ray and neutron-scattering techniques (known as SAXS and SANS nrespectively) are low resolution techniques that can characterize protein structure in solution. They are of particular utility when large proteins cannot be crystallized and in systems where solution conditions affect the structure adopted. Atomistic models of the average structure can be generated through constrained modelling, a technique in which known structures (such as domain or subunit crystal structures) are combined with linker models to produce candidate global conformations. Homology modelling techniques and simulation methodologies such as Monte Carlo or molecular dynamics simulations can be used to produce a library of structurally varied, atomistic, candidate models of the target macromolecule. Theoretical scattering curves are then generated for each model and used for trial-and-error fits to the experimental data. From this a small family of best-fit models can be identified. It is to automate this last two steps that SCT was developed. The original SCT software, written in Fortran, has been used in the production of 74 structures (21 antibodies, 27 complement proteins and 24 oligosaccharides) deposited in the Protein Data Bank between 1998 and 2014. All of the same functionality of the classic SCT is now available in an easier to use set of Python scripts.
Features
- Creates low resolution sphere models from atomistic models (input in PDB format)
- Addition of hydration layers to models for comparison to SAXS data
- Calculation of scattering curves from sphere models (via the Debye equation)
- Computation of R factor or Chi squared metrics of the comparison between experimental and modelled curves
- Calculation of sequence based estimates of protein volume
- Python package facilitating the develoment of new workflows
This software was developed in the Structural Immunology Group at UCL. The original developer was Professor Stephen J Perkins (s.perkins@ucl.ac.uk) and the Python version was created by David W. Wright (dave.wright@ucl.ac.uk).